

 Share
Share Tweet
Tweet Share
Share
5 Promising Applications of Machine Learning in Drug Discovery

29 February 2024
Welcome to the future of pharmaceuticals, where the fusion of science and technology opens new horizons in healthcare. Imagine a world where the journey from the laboratory to the patient’s bedside is faster, more efficient, and more precise. This is not a glimpse into a distant future; it’s the reality we’re stepping into today, thanks to the groundbreaking integration of Machine Learning (ML) in drug discovery.
Curious about the intersection of Machine Learning and Life Sciences? Learn how Shiny applications are unlocking new possibilities.
In this blog post, we embark on an exhilarating exploration of how Machine Learning is transforming the landscape of drug discovery. The quest for new medications has always been a complex and challenging one, fraught with uncertainties and demanding immense resources. However, the advent of ML is reshaping this landscape, turning what was once science fiction into a tangible reality.
Table of Contents
- Target Identification and Prioritization
- Protein Structure Prediction
- Binding Affinity Prediction
- Drug Repurposing
- De-novo Drug Design
- Take The Next Step
Target Identification and Prioritization

Drug Target Identification
Target identification in drug discovery involves finding biological targets, like proteins or genes, crucial in diseases. These targets are validated to confirm their role in the disease and potential as drug targets. Post-validation, targets are prioritized for drug development based on their disease role, druggability, and patient impact.
Return on Investment (ROI)
Target discovery done experimentally is difficult to carry out quickly and widely due to limitations in throughput, accuracy, and cost. The use of ML at this stage can reduce the time to identify and prioritize a target and reduce the time required from years to days.
Open Targets
An example of ML being applied in this task is Open Targets. This innovative, large-scale, multi-year, public-private partnership uses human genetics and genomics data for systematic drug target identification and prioritization.
The Open Targets Platform integrates public domain data to enable target identification and prioritization.

Open Targets Platform
The Open Targets Genetics is a comprehensive tool highlighting variant-centric statistical evidence to allow both the prioritization of candidate causal variants at trait-associated loci and the identification of potential drug targets.
Protein Structure Prediction
The obtention of a 3D structure of a large biomolecule (target) is the next step once a target has been identified. This 3D representation aids the medicinal chemist in performing the hit-to-lead optimization through a rational, structurally enabled process to find the optimized candidates that strongly and optimally bind to the desired target site. The challenge lies in the fact that acquiring 3D structures of protein-ligand complexes can be complex, typically requiring methods like X-ray crystallography and cryo-electron microscopy (cryo-EM).
ROI
Speed is a big deal in drug discovery. Determining the 3D structure of a protein usually takes from many months to years; with the use of AI models, such as AlphaFold, it now takes seconds. In some cases, an AlphaFold structure could jump-start a project and eliminate years of work in trying to obtain a new structure.
On the other hand, having an experimental structure continues to be more reliable, especially considering that protein structure varies and is not rigid when bound to a drug. X-ray crystallization is still considered the gold standard for obtaining new 3D structures of macromolecules such as proteins. Estimates are that for drug discovery, there is a 50% cost and time savings from target selection to IND filing when structure is centrally used as part of the drug discovery process.
AlphaFold
AlphaFold is an AI system developed by Google’s DeepMind that predicts a protein’s 3D structure from its amino acid sequence. It was taught by showing the sequences and structures of around 100,000 known proteins. This AI system can now predict the shape of a protein almost instantly.
The AlphaFold DB provides open access to over 200 million protein structure predictions.

AlphaFold
The latest AlphaFold model significantly improves accuracy and expands coverage beyond proteins to other molecules, including ligands.
Using Machine Learning to Identify Protein Crystals
There are many limitations in obtaining 3D structures of targets experimentally. This can be solved with machine learning.
We addressed this issue through machine learning; utilizing a dataset commonly used by major pharmaceutical companies (MARCO dataset), we developed a model to detect protein crystals more accurately and established a new state-of-the-art (SOTA).
A New SOTA
Our method, which involves using pre-existing models and specialized binary classification, surpasses the advanced MARCO model by lowering the chance of overlooking crystals from 11.1% to 7.6% in MARCO’s test dataset. This signifies a more than 30% reduction in the initial error rate.
Moreover, we are able to achieve superior performance on new data using as few as 60 images of crystals.
Important Metrics:
- Crystal Identification Accuracy:
- Old Method (MARCO): 88.9% recall and 93.4% precision on crystals. [*]
- New Method: 90.8% recall and 93.9% precision on crystals (4-class model), and a recall of 92.4% with a precision of 93.4% (binary classification model).
- Overall Accuracy:
- Old Method: 93.5% overall accuracy for 4-class classification, 97.7% for detecting crystals.
- New Method: 94.0% overall accuracy (4-class model) and 98.1% (binary classification model).
- Fine-tuning to new data:
- Old Method: on new “VIS” and “UV” datasets, MARCO achieves crystal detection accuracy of 91.1% and 75.7%, respectively.
- New Method: fine-tuned using as few as 60 images of crystals, our model obtains accuracies of 92.9% and 81.1%, respectively; using 2000 images of crystals, the accuracy increases to 95.7% and 91.9%, respectively.
- Computational Effort:
- Old Method: 260 training epochs across 50 GPUs.
- New Method: 3 + 2 training epochs (4-class model + binary model) on a single GPU.
This breakthrough has the potential to provide substantial cost and time savings in drug development.
Learn more about our protein crystal detection project in this blog post – Accelerating Drug Discovery: Machine Learning for Protein Crystal Detection.
Binding Affinity Prediction
Protein Complex and Inhibitor Molecule
Computer-Aided Drug Design (CADD) greatly benefits from the ability to predict the binding affinity (BA) between a protein and a potential drug candidate. This capability is crucial for identifying new small molecules from costly compound libraries. Binding affinity is used to comprehend a drug’s effect on the body. Ligands that bind strongly to a target protein are chosen as drug candidates.
ROI
Accurately predicting a ligand’s binding affinity can lower the expenses associated with de novo drug design by enhancing the prediction’s precision when used for virtual screening or lead optimization.
DeepDTA
The DeepDTA is a deep-learning based model that uses only sequence information of both targets and drugs to predict DT (drug-target) interaction binding affinities. The approach followed by this project is the modeling of protein sequences and compound 1D representations (SMILES) with convolutional neural networks (CNNs) to predict the binding affinity value of drug-target pairs. The GitHub repository can be found here.
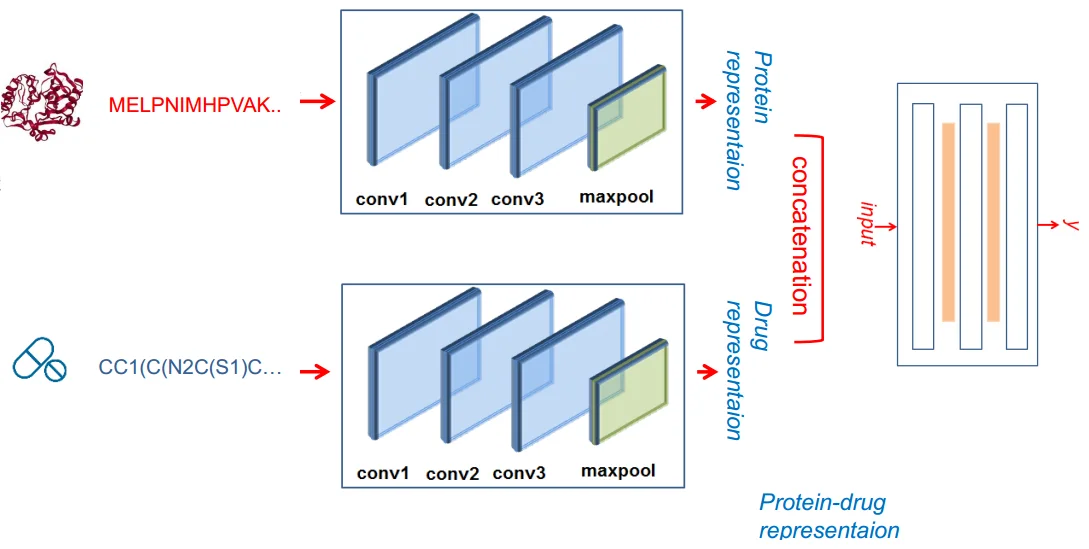
deepDTA Model Architecture
BiComp-DTA
BiComp is a unified measure for protein sequence encoding. It provides compression-based and evolution-related features from the protein sequences, specifically, normalized compression distance and smith-waterman measures for capturing complementary information from the algorithmic information theory and biological domains, respectively. This new model was evaluated using four benchmark datasets for drug-target binding affinity prediction and provided superior efficiency in terms of accuracy, runtime, and the number of trainable parameters. This method can be run on a normal desktop computer.
Learn how wearables and AI are revolutionizing assessments in musculoskeletal health. Visit our blog post for more details.
Drug Repurposing

Drug Repurposing
In treating diseases, there are two main strategies: de-novo drug design and drug repurposing. De-novo drug design begins with studying the structure of protein receptors, typically explored using methodologies like x-ray crystallography or Cryo-EM. On the other hand, drug repurposing involves exploring new effects of existing drugs or drug combinations. The first strategy, de novo design, typically requires billions of dollars, further, bringing a new drug to the market takes between 10 to 15 years.
ROI
Drug repurposing reduces the time and cost of drug development; additionally, since these drugs have already been explored in clinical trials and are proven to be safe in humans, Phase I trials can be skipped. A significant advantage of this approach is the ability to provide effective treatments to patients in need more rapidly.
Another significant benefit is the increased return on investment for pharmaceutical firms striving to launch new medications, as this method is more cost-effective compared to traditional drug discovery processes. Utilizing drug repurposing can cut down costs to under half a billion dollars, markedly influencing the research and development (R&D) budget of a company.
Key Principles
Through widespread experimentation, two key principles have been established for the logical structuring of drug repurposing.
The first principle highlights the diverse characteristics of drugs. While drugs are typically crafted to attach to a particular target, they frequently engage with various targets or pathways upon entering the body, a phenomenon often referred to as the off-target effect.
The second key principle concerns the adaptability of the target. The onset of complex diseases is often linked to the irregular expression of various genes. Extensive studies on disease-gene networks have revealed numerous interactions between drugs and their targets, indicating that a target linked to a specific disease or pathway might also play a role in other disease mechanisms. Consequently, a drug acting on such a target could be effective against multiple diseases. This multifaceted nature of drug targets suggests that the possible uses of established drugs can be forecasted by identifying common target molecules in different diseases. This concept has led to the successful repositioning of several known drugs.
The Kuala Framework
The KUALA framework stands out by automating the identification and ranking of kinase active ligands, focusing on the best candidates for repurposing. It brings to light the substantial similarity in kinase binding sites, which is pivotal for drug selectivity and poly-pharmacology.
DrugSolver CavitomiX
This tool, accelerated by GPU on NVIDIA DGX, specializes in drug repositioning and identifying off-target effects using cavity property point clouds. It employs these point clouds to examine enzymes that have analogous binding sites, regardless of their structural or sequential differences. The method is versatile, applicable to biocatalysis, the discovery of drugs, reevaluating existing drugs, and detecting off-target interactions.
De-novo Drug Design

Drug Design
De novo drug design is an approach that generates new chemical entities solely from knowledge about a biological target (receptor) or its identified active binders. These new chemical entities will ideally possess desirable properties such as efficacy, safety, and PK/PD (pharmacokinetic/pharmacodynamic) profiles.
Identifying new chemical entities that exhibit the required biological activity is essential to sustain the drug discovery pipeline. Consequently, creating innovative molecular structures for synthesis and in vitro testing is imperative in developing new treatments for future patients.
Despite recent advances, limited exploration of chemical space for new drugs poses significant challenges for chemists in developing novel molecules for drug discovery.
Explore how our Drug Interactions Shiny App illuminates pharmacokinetic and pharmacodynamic interactions — visit our blog post to learn more.
ROI
Transforming a chemical entity into a marketable drug through its development, testing, evaluation, and approval is a demanding and costly endeavor with a high risk of failure. AI-assisted de novo design enables the creation of a broader range of structures, eliminating the reliance on pre-existing compound libraries.
An example of the return on investment that could be brought by applying AI/ML at this stage of the discovery process is shown by Insilico Medicine. This AI drug discovery company succeeded in using de novo AI to design a new molecule in just 21 days and further validate it in only 25 days; this represents an acceleration of de novo discovery of 15 times.
REINVENT 2.0
This is an AI tool for de novo drug design. The tool can be effectively applied to drug discovery projects that are striving to resolve either exploration or exploitation problems when navigating the chemical space. It aids chemical structure idea generation by displaying the most promising compounds. REINVENT’s code is publicly available at https://github.com/MolecularAI/Reinvent.
Take The Next Step
Interested in harnessing the power of machine learning for your drug discovery projects? You’re at the right place. Our expertise in integrating machine learning with pharmaceutical research can propel your projects to new heights of innovation and efficiency.
Don’t let the potential of AI in drug discovery remain untapped for your business. Contact us today.
Eager to upgrade your R/Shiny proficiency for Life Sciences? Grab our free eBook and start advancing your skills today.

Contact Us

Head of Sales

Get Updates
Subscribe to Shiny Weekly Newsletter
Join 4000+ Shiny enthusiasts to see the latest Shiny news from the R community.
Also on drug discovery
